PI: John M. Essigmann, Departments of Chemistry, Biological Engineering and Center for Environmental Health Sciences, MIT
Abstract
Hepatocellular carcinoma (HCC) is the second leading cause of cancer death worldwide, responsible for over 700,000 deaths each year. Despite ample epidemiological evidence for multiple chemical and viral risk factors, and opportunities to mitigate such risks, HCC remains a particularly deadly disease because it is asymptomatic until the disease reaches a late stage. These considerations highlight the need for early detection of the mutagenic processes that initiate and/or promote carcinogenesis in the liver. Early detection could lead to surgical intervention, which would lead to decreased mortality. Moreover, biomarkers that truly reflect the mutagenic mechanisms that cause or promote the disease can be used to evaluate chemo-prevention strategies. This proposal uses high resolution mutational fingerprinting to identify liver cancer before clinical disease is evident. We have proven the technology with one cause of liver cancer (aflatoxin) and propose to apply it to determine the signature of liver cancer induced by alcohol
Report
- Project Title: Early onset biomarkers enable intervention against liver cancer
- Principal Investigator: John M. Essigmann, William R. (1956) & Betsy P. Leitch Professor in Residence Professor of Chemistry, Toxicology, and Biological Engineering
- Grant Period: September 2017 – August 2018
Summary
This is a progress report on a Seed Project funded by the Skoltech-MIT Fund. The project is a few months from completion but most of the important parts of the work are done. Overall, this project exploits a recently developed DNA sequencing technique to look for the patterns of mutation that occur in mouse liver after the mouse has been exposed to a two-stage regimen of ethyl alcohol. In the first stage, mice are maintained on a 5% ethanoldiet (this is approximately the concentration of ethanol in beer), followed by a binge dose of ethanol (roughly equivalent to one-half to two-thirds of a bottle of hard distilled spirits). This regimen models the behavior of humans who drink both chronically (i.e., every day) with occasional heavy episodes of binge drinking. Both males and females were evaluated.
Background
Epidemiological studies have firmly established ethanol as a risk factor for human cancers including hepatocellular carcinoma (HCC) (1). Liver diseases associated with alcohol consumption range from transient steatosis to steatohepatitis, cirrhosis and hepatocellular carcinoma (2). Acute damage from binge exposure can trigger apoptosis and necrosis, stimulating regenerative responses involving mutagenic DNA replication (3, 4). While the metabolic pathways involved in the pathogenesis of alcohol-induced liver disease have been well studied, less information is available about their roles as mediators of genetic damage in promoting liver cancer.
Liver cancer is the second leading cause of cancer deaths worldwide, following only lung cancer in global mortality (5). The four leading causes of HCC are the food contaminant aflatoxin B1(AFB1), alcohol, and hepatitis B and C viruses; alcohol and hepatitis C are the leading hepatocarcinogens in Russia. In a recent project we used a high-fidelity duplex DNA sequencing (DS) technique to reveal a distinctive, high-resolution mutational fingerprint of AFB1in a mouse model. The fingerprint appeared very early during the development of the disease, leading to speculation that, in liquid biopsy samples, it could be used as abiomarker to afford detection of cancer at a point at which surgical intervention would still be possible (6). One goal of the work described here is the use DS analyses to identify the mutational mechanisms responsible for ethanol-induced genetic changes leading to HCC. The AFB1mutational fingerprint in our mouse model recapitulated a major subset of human HCCs. In the work on ethanol, we are taking the same approach to determine the mutagenic fingerprints and associated mechanisms underlying induction of HCC by ethanol.A second goal of our project was to use a systems biology approach in collaboration with investigators at Skoltech, MIT and the University of Pennsylvania to identify the key biochemical changes responsible for ethanol-induced genotoxicity in liver.
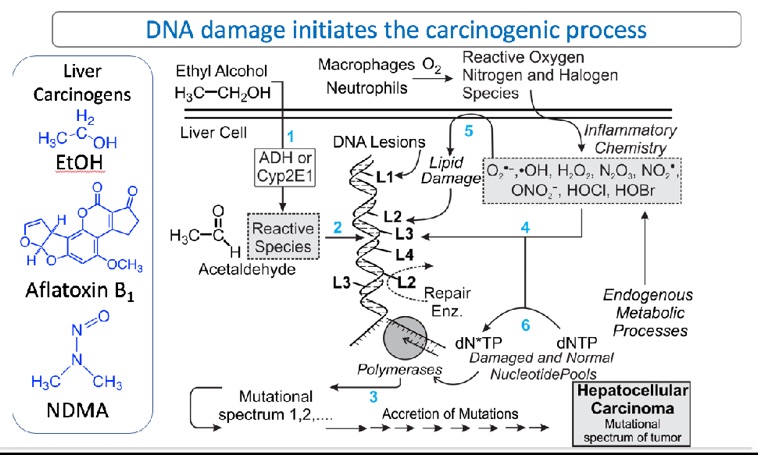
Fig. 1. Recent work showing high-resolution mutational spectra of mouse liver at various points in time after administration of the liver carcinogen, AFB1. The proposed workflow will be similar, with substitution of ethanol (and ethanol plus positive control NDMA), for AFB1as the liver carcinogen. Details are in Chawanthayatham et al. (2017).
Figure 1 shows the framework for the project. Ethanol enters the cell (step 1) and is metabolized by Phase I enzymes (such as cytochrome P4512 E1) to electrophilic intermediates, thefore most of which is acetaldehyde. Such electrophiles react with nucleophilic sites on DNA to form covalent metabolite-DNA base adducts (step 2). One of these adducts is N2-ethylguanine. In addition to production of organic DNA adducts, ethanol induces inflammation, which is associated with production of the reactive oxygen, nitrogen and halogen species shown in the box of Figure 1; these agents also damage DNA, forming oxidized and halogenated DNA bases such as 7,8-dihydro-8-oxoguanine and 5-chlorocytosine, respectively. These adducts formed following ethanol exposure can cause replication errors resulting in mutations (step 3). Collateral damage of membranes and nucleotide pools (steps 4-6) also results in the formation of electrophiles that form additional DNA lesions. All of aforementioned DNA adducts are mutagenic and could be drivers of the genetic changes that trigger the conversion of normal liver cells to liver cancer cells, which outgrow into a tumor.
Progress
Our studies employed a unique genetically engineered mouse strain containing a lambda transgenic mutational reporter (the gpt-delta C57BL/6 mouse) (6).
The animals were dosed with ethanol as indicated in Figure 2. Briefly, ten-week old male and female mice were acclimated over five days to a liquid Lieber-DeCarli diet and then split into two groups. One group was fed diet containing 5% ethyl alcohol for 10 days and the other, control, group was fed the diet without ethanol. After ten days, the ethanol group was administered a binge (5g/kg) dose of ethanol by gavage. Five days later the animals were sacrificed.
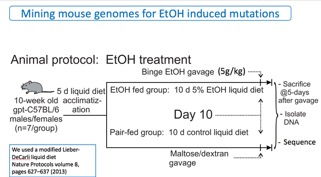
Fig. 2. Protocol for dosing mice with ethyl alcohol (EtOH).
At the termination of the experiment, samples were taken for histology and RT-PCR to look for certain biomarkers of animal health status. A portion of the liver was taken by a Skoltech student of Prof. Timofei Zatsepin; she isolated DNA from the liver tissue and returned with it to Russia for analysis of DNA lesions formed after ethanol treatment. Blood plasma was taken to be shipped to Prof. Ian Blair at the University of Pennsylvania School of Medicine. Blair is an expert in the use of mass spectrometry to determine inflammatory cytokines released following exposure of tissues to toxicants. He specifically is examining the blood for evidence of HMGB1 release. Another portion of the tissue was given to Prof. Steven Tannenbaum, who is using mass spectrometry to determine the changes in the metabolome in response to ethyl alcohol. Ethanol is expected to change the relative amounts of intracellular metabolites, and metabolomics is the proper type of analysis to observe these pathwaydisruptions.
Approximately 25 mg of liver tissue was taken for mutational analysis, which is the specialty of our laboratory. The genome of the gpt-delta C57BL/6 mouse contains ~80 copies of a 6.4 kb target that we deep sequence using the DS technique. By this method, we sequence each base ~15,000 times. Prior to sequencing, the two strands of DNA are tagged with a barcoded adapter, separated, and each strand is sequenced independently. Post-sequencing, sequencing errors that occur in one strand (e.g., the Watson strand) but not in the Crick strand are computationally filtered out. Only true mutations that occur in both the Watson and Crick strands are retained. This technique is ~104times more accurate than conventional next generation sequencing.
The animal portion of the experiment is almost done; tissues have been distributed to collaborators and we are in the process of isolating and sequencing DNA. That process will take several months. After sequencing, the data are transmitted from the sequencing center to our laboratory and put into the hands of our bio- informaticist. She assembles the mutational spectrum in which a mutant base is presented in the context of its two neighbors (the 5’ and 3’ non-mutant bases that flank a mutated site). There are six possible base substitution mutations and a total of 16 (4x4) three-base contexts, resulting in a 96-bin spectrum (4x4x16=96). This spectrum is then computationally compared to the repository of approximately 300 liver cancers for which we have good sequencing data. Lastly, we identify human tumors that have mutational spectra that are similar to the one produced by ethanol in the mouse based on a statistical measure (cosine similarity). People whose mutational spectra have cosine similarity close to that of the mouse are the people we predict have had liver cancers that could have alcohol abuse in their etiology. Once the mutation data are in and processed, we shall meet with our Russian and other collaborators to determine a publication strategy.
Perspective
This Seed Project might provide data similar to those we obtained with aflatoxin, where a biomarker of eventual liver cancer appeared only ten weeks after administration of a single dose of the carcinogen. Aflatoxin is a powerful liver carcinogen, and mutagen, so we accept the possibility that a weaker carcinogen such as ethanol might leave a weaker predictive biomarker. Nevertheless, this project is a good beginning and it is fortunate that other investigators have joined, which gives us several shots on goal to find metabolic and genetic markers that could predict later-life cancer induction. The importance of early biomarkers stems from the sad reality that liver cancer is untreatable by chemo-or immune-therapy. Prevention and early diagnosis are the best current approaches to mitigate this disease. In our work, we hope to detect the disease at a point in time when the tumor can be surgically removed. While prevention is always the best cancer mitigation strategy, surgical removal of the tumor is an excellent secondoption.
Literature Cited
-
Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma.Gastroenterology. 2004;127(5 Suppl 1):S87-96. PubMed PMID:15508108.
-
Stewart S, Jones D, Day CP. Alcoholic liver disease: new insights into mechanisms and preventative strategies. Trends Mol Med. 2001;7(9):408-13. PubMed PMID:11530336.
-
Brandon-Warner E, Schrum LW, Schmidt CM, McKillop IH. Rodent models of alcoholic liver disease: of mice and men. Alcohol. 2012;46(8):715-25. PubMed PMID: 22960051; PubMed Central PMCID: PMC3496818.
-
Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013;8(3):627-37. PubMed PMID: 23449255; PubMed Central PMCID: PMC3788579.
-
WHO. WHO Factsheet: WHO; 2017 [cited 2017]. Available from:http://www.who.int/mediacentre/factsheets/fs297/en/.
-
Chawanthayatham S, Valentine CC, 3rd, Fedeles BI, Fox EJ, Loeb LA, Levine SS, Slocum SL, Wogan GN, Croy RG, Essigmann JM. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2017;114(15):E3101-E9. PubMed PMID: 28351974; PubMed Central PMCID:PMC5393230.